Introduction¶
“Multiplexing” is a sequencing technique, which pools many samples in the same sequecning batch, where samples are distinguished by barcode sequences. It helps enhance throughput, optimize cost, and simplifiy analysis Sample Multiplexing | Multiplex Sequencing with Indexes, 2026. In contrast, “demultiplexing” resolves that which sample that sequencing read belonging.
The command in the tutorial
qiime demux emp-single \
--i-seqs emp-single-end-sequences.qza \
--m-barcodes-file sample-metadata.tsv \
--m-barcodes-column barcode-sequence \
--o-per-sample-sequences demux.qza \
--o-error-correction-details demux-details.qzaCommand explanation:
Inputs:
--i-seqs: Including fastq files of barcode and sequencing read.--m-barcodes-file: The metatdata contains auxiliary information according to barcode.
Parameters:
--m-barcodes-column: The column in the metadata file that contains the barcode sequences.
Outputs:
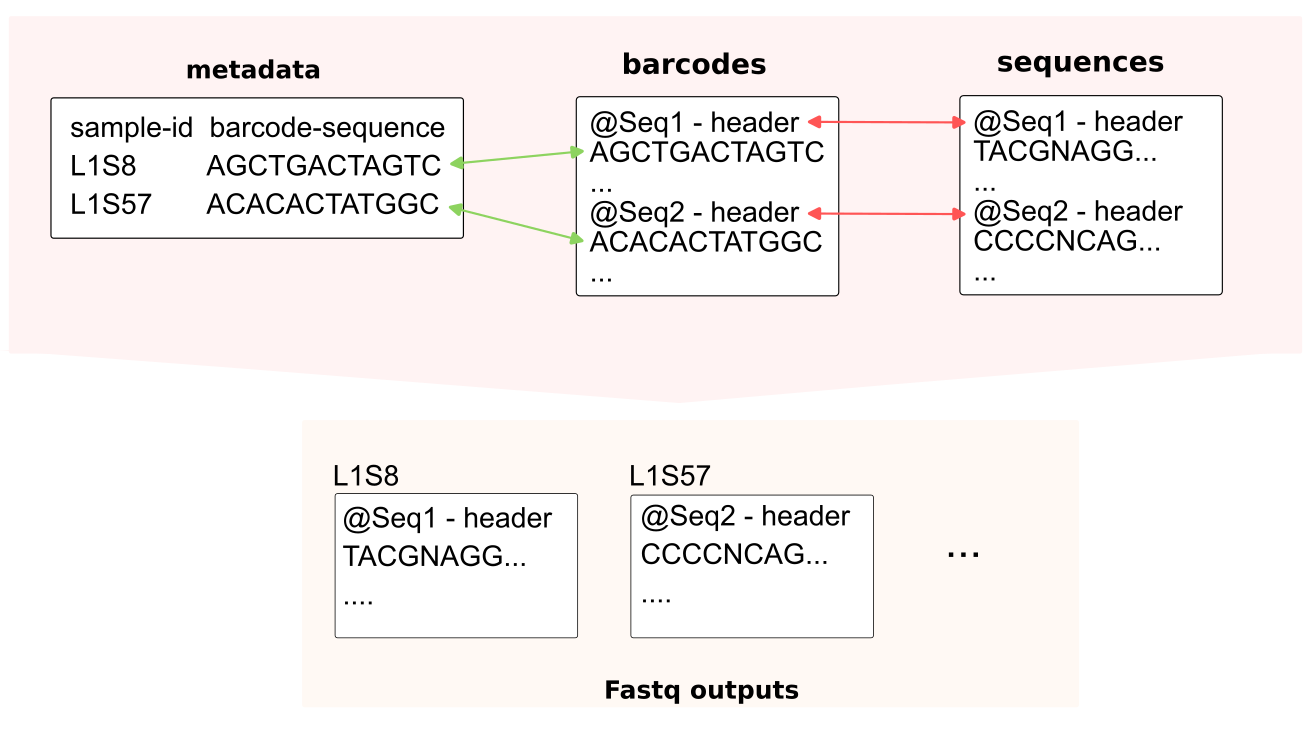
The ultimate purpose is generating fastq files of each sample from a mixed fastq file. Base on the sequencing header on a read (in sequences fastq file), the original sample (in metadata) is retrieved by the barcode associating the same the sequencing header (in barcodes fastq file).
Demultiplexing locks in downstream accuracy. Within demux emp-single, the process relies on rigid defaults that cause silent data loss if core assumptions are violated, such as unexpected barcode orientations, non-Golay formats, sequencing errors exceeding 3 bits, or altered barcode lengths. Because these issues directly compromise sample-to-read mapping, looking under the hood is essential to diagnose and monitor data attrition.
Demultiplex internal steps¶
Sequencing reads are processed one by one from the input FASTQ file. For each read, the associated barcode sequence is retrieved. Although, the step “Reverse complement barcode” was not performed in this command (demux emp-single) by default, more information can be found in Reverse complement barcode.
Next, Golay error correction is applied by default to the barcode, allowing correction of sequencing errors in barcode sequence (up to three mismatches) and improving robustness in sample identification (read more in Golay error correction).
The corrected barcode is then matched against a predefined barcode-to-sample mapping. If a match is found, the corresponding read is assigned to that sample and written to its output FASTQ file. Reads that do not match any known barcode are discarded.
Over the course of processing, reads are thus separated into multiple per-sample FASTQ files, each representing an individual sample from the original multiplexed dataset.
Reverse complement barcode¶
Reverse complementing is required when barcode sequences in the reads are in the opposite orientation to those in the metadata mapping file, which can occur depending on sequencing setup (e.g., Illumina index reads). If not corrected, this mismatch can result in failed or low-rate sample assignment.
QIIME 2 provides two options to handle this:
--p-rev-comp-barcodes: which activate reverse complement step in the demux workflow. Barcode sequences will be reverse complemented. Use this when the barcodes in file are misaligned to the barcodes in the metadata file.--p-rev-comp-mapping-barcodes: in constrast, reverse complements the barcodes in the metadata mapping file.
Only one option should be used, depending on which side has correct orientation.
Golay error correction¶
Golay error correction is an error-correcting coding method that represents DNA barcodes as structured 24-bit vectors, enabling detection and correction of sequencing errors based on the properties of the Golay (24,12,8) code Morelos-Zaragoza, 2006, pg.30-31. It is used in sequencing workflows to improve sample assignment accuracy. Because sequencing errors are common, exact barcode matching can lead to substantial data loss.
Golay coding allows correction of maximal three bit errors, depending on type of correction, the number of bit changed is different. If the number of bit errors exceeds three, the record is discarded.
| Correction | Number of bit changed |
|---|---|
| A T | 1 |
| C G | 1 |
| {A,T} {C,G} | 2 |
Correction workflow:
Convert barcode (DNA) to a 24-bit vector (2 bits per nucleotide)
Compute the syndrome using a parity-check matrix (H)
Use the syndrome to identify the most likely error pattern (lookup table)
Correct the bit vector by applying the error pattern
Convert the corrected bits back to a DNA sequence
Golay error correction assumes that true barcodes belong to the predefined set of valid Golay codewords ( valid barcodes), that barcodes have a fixed length of 12 nucleotides (corresponding to 24-bit codewords), that sequencing errors are limited (typically no more than three bit errors), and that these errors occur randomly rather than systematically.
Full python script for Golay error correction can be found in GolayDecoder.py.
Summary¶
To summarize, default settings should not be trusted when using non-standard protocols (e.g., non-Golay barcodes, different barcode lengths, or unknown orientation), or when preprocessing steps may have inadvertently mixed up the barcode sequences.
- Sample Multiplexing | Multiplex Sequencing with Indexes. (n.d.). Retrieved April 14, 2026, from https://www.illumina.com/techniques/sequencing/ngs-library-prep/multiplexing.html
- Morelos-Zaragoza, R. H. (2006). The Art of Error Correcting Coding (2nd ed). John Wiley.