Introduction¶
Similar to DADA2, Deblur is a denoising method designed to correct errors occurring during sequencing and PCR amplification cycles, which otherwise limit the ability to perform fine-scale classification in 16S rRNA amplicon sequencing Amir et al., 2017.
Instead of using traditional Operational Taxonomic Units (OTUs) or Amplicon Sequence Variants (ASVs) of DADA2, Deblur introduced a novel approach termed Sub Operational Taxonomic Units (sOTUs).
There are two main steps in Deblur pipeline according to two commands mentioned in the tutorial.
The command in the tutorial
The command for preparation by quality filtering:
qiime quality-filter q-score \
--i-demux demux.qza \
--o-filtered-sequences demux-filtered.qza \
--o-filter-stats demux-filter-stats.qzaCommand explanation:
Inputs:
--i-demux: The input demultiplexed sequences artifact. Note: Output from Demultiplexing.
Outputs:
--o-filtered-sequences: The resulting quality-filtered sequences artifact. Note: Used as input for the next Deblur step.--o-filter-stats: A summary of the quality filtering results.
And the deblur command:
qiime deblur denoise-16S \
--i-demultiplexed-seqs demux-filtered.qza \
--p-trim-length 120 \
--p-sample-stats \
--o-representative-sequences rep-seqs-deblur.qza \
--o-table table-deblur.qza \
--o-stats deblur-stats.qzaCommand explanation:
Inputs:
--i-demultiplexed-seqs: The input quality-filtered sequences artifact. Note: Output of the quality filtering step.
Parameters:
--p-trim-length: The fixed length to which all sequences are trimmed.--p-sample-stats: A flag to request per-sample statistics.
Outputs:
--o-representative-sequences: The resulting sequences for each sOTU.--o-table: The resulting feature table.--o-stats: Detailed statistics for the Deblur process.
Workflow¶
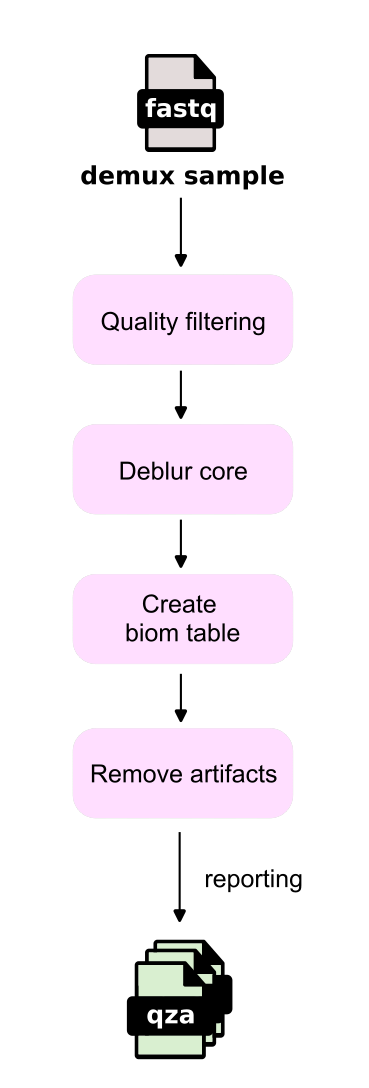
The workflow starts with demultiplexed FASTQ files representing each sample, with each sample undergoing the Deblur workflow separately. Each record first passes through Quality filtering, which involves truncation based on quality scores.
Subsequently, the Deblur core process is performed to determine sOTUs and remove chimeric sequences (sequences likely belonging to two parents). Finally, a BIOM table is created (Create biom table) for downstream analysis, ensuring it is free from the noise of artifacts (Remove artifacts).
Quality filtering¶
The quality filtering process is performed independently for each sample and consists of three main steps: (1) scanning each read for a low-quality window, (2) truncating the FASTQ record based on the position of that window, and (3) tracing and labeling each record based on the results of the filter.
A low-quality window is identified as the first instance of consecutive bases with Phred quality scores below a specific threshold (the default is 4). Once this window is found, the read is truncated at that position. Each read is then assigned a status label to track its filtering outcome:
| Label | Meaning |
|---|---|
| untruncated | The read was not truncated; the quality remained above the threshold throughout; do not contains ambiguous bases (N) |
| truncated | The truncated read has accepted truncated fraction, and do not obtain ambiguous bases |
| short | The read has truncated fraction (truncated length / read length) is greater than 0.75 |
| ambiguous | The read without being truncated obtains ambiguous bases |
| truncated ambiguous | The truncated read obtains ambiguous bases |
Deblur core¶
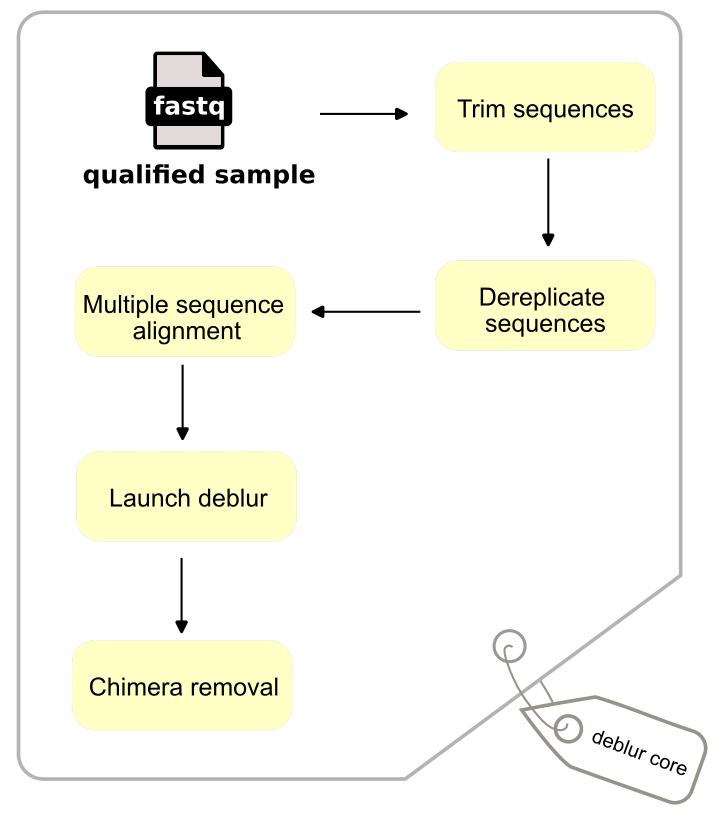
Qualify sequences are trimed to be equal in length (120 bp), sequences have length being shorter than the trim length are discard. Deprelication is performed by counting abundance of unique sequences and removing singletons using VSEARCH (v2.22.1) Rognes et al., 2016.
Following standardization, sequences were subjected to multiple sequence alignment (MSA) using MAFFT Katoh & Standley, 2013. To accommodate the large-scale nature of the dataset, the PartTree algorithm was employed; this approach reduces the complexity of pairwise comparisons to by recursively partitioning sequences based on their similarity to a subset of ‘seed’ sequences Katoh & Toh, 2007.
Metaphorically, Launch Deblur initializes a competition where each sequence is a competitor, and their “mana” represents their observed abundance. Starting with the most abundant, sequences duel one by one until the end, the survivors remain as sOTUs. In each match, the “damage points” dealt by a sequence to another represent the statistical number of error copies expected if the opponent were merely its own sequencing artifact (empirical algorithmic code can be found in deblur.py).
The Chimera removal step is performed by VSEARCH using the UCHIME de novo algorithm Edgar et al., 2011. This approach operates on the assumption that ‘parent’ sequences coexist in the same FASTQ file as their chimeric artifacts. Any sequence with an abundance exceeding a specific threshold (default is 2) is considered a potential parent and stored in a local reference set. The algorithm processes sequences in order of decreasing abundance, if a query sequence is found to be a significant match-constructed from a combination of two parents in reference, it is flagged as a chimera and discarded.
Create biom table¶
The denoised output from the Deblur core process across all samples is compiled into a single BIOM table. This table consists of a matrix (dimensions: ) where the cell values represent the remaining frequency (abundance) of each sequence. Note that samples containing zero reads are excluded from the matrix.
Additionally, any sOTUs with a total cross-sample abundance falling below a specified threshold (default is 10) are discarded to filter out rare artifacts. Both the final BIOM table and the corresponding sOTU sequences are preserved for downstream analysis.
Remove artifacts¶
SortMeRNA v2.0 Kopylova et al., 2012 is employed to remove artifact sequences, which by default are composed of PhiX and sequencing adapters , see artifact.fa.
Artifact sequences that match the references are stored separately from the sOTU sequences. Any samples that become empty after this filtration are discarded, and the BIOM table and sequence file are updated accordingly.
Summary¶
Overall, Deblur’s strategy focuses on removing likely error sequences, which are minor and significantly different from others, by subtracting their abundance from neighboring sequences. This leads to a drop in the observed frequency of these noise reads. In contrast, DADA2 accounts for these errors by incorporating their abundance into the figures of their respective ASVs (Amplicon Sequence Variants).
Regarding performance, Deblur is generally slower than DADA2. This is because Deblur processes each sample and record sequentially and is primarily written in Python. Conversely, DADA2 utilizes parallel processing for steps such as Trim & Filtering records, and chimera removal. Moreover, its core algorithm executed via a high-performance C++ API.
The Quality filtering step in Deblur focuses on truncation and the removal of low-quality windows, though the reason for specific labeling has not been understood yet. Another key distinction is that Deblur assumes the parents of chimeras coexist within the same FASTQ file and dominating when removing chimeras, whereas DADA2 performs “chimera voting” across multiple samples to increase accuracy.
- Amir, A., McDonald, D., Navas-Molina, J. A., Kopylova, E., Morton, J. T., Zech Xu, Z., Kightley, E. P., Thompson, L. R., Hyde, E. R., Gonzalez, A., & Knight, R. (2017). Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems, 2(2), e00191-16. 10.1128/mSystems.00191-16
- Rognes, T., Flouri, T., Nichols, B., Quince, C., & Mahé, F. (2016). VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ, 4, e2584. 10.7717/peerj.2584
- Katoh, K., & Standley, D. M. (2013). MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Molecular Biology and Evolution, 30(4), 772–780. 10.1093/molbev/mst010
- Katoh, K., & Toh, H. (2007). PartTree: An Algorithm to Build an Approximate Tree from a Large Number of Unaligned Sequences. Bioinformatics, 23(3), 372–374. 10.1093/bioinformatics/btl592
- Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., & Knight, R. (2011). UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics, 27(16), 2194–2200. 10.1093/bioinformatics/btr381
- Kopylova, E., Noé, L., & Touzet, H. (2012). SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics, 28(24), 3211–3217. 10.1093/bioinformatics/bts611