Introduction¶
In 16S rRNA analysis, the first step in characterizing the microbiome of a sample is to cluster similar sequencing reads into representative units that can be used for taxonomic assignment. However, sequencing errors can introduce false positives, leading to incorrect biological interpretations. Traditionally, this clustering is performed by grouping reads into Operational Taxonomic Units (OTUs) based on a fixed sequence dissimilarity threshold. Nevertheless, this approach often lacks sufficient resolution to capture fine-scale taxonomic variation and can result in misinterpretation of microbial diversity Callahan et al., 2016.
DADA2 address this issue by attempting to correct sequencing errors at the single-nucleotide level without constructing OTUs. Instead, they generate Amplicon Sequence Variants (ASVs) Nearing et al., 2018.
The command in the tutorial
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences rep-seqs.qza \
--o-table table.qza \
--o-denoising-stats denoising-stats.qza \
--o-base-transition-stats base-transition-stats.qza
Command explanation:
Inputs:
--i-demultiplexed-seqs: The input artifact containing demultiplexed sequences. Note: Output from Demultiplexing.
Parameters:
--p-trim-left: The number of bases to remove from the start (5’) of the sequence.--p-trunc-len: The position at which sequences should be truncated.
Outputs:
--o-representative-sequences: The resulting sequences for each ASV. Note: Used for Phylogenetics.--o-table: The resulting feature table (ASV table). Note: Used for Diversity analysis.--o-denoising-stats: A summary of denoising results.--o-base-transition-stats: Statistics on error rates estimated during the learning phase.
Workflow¶
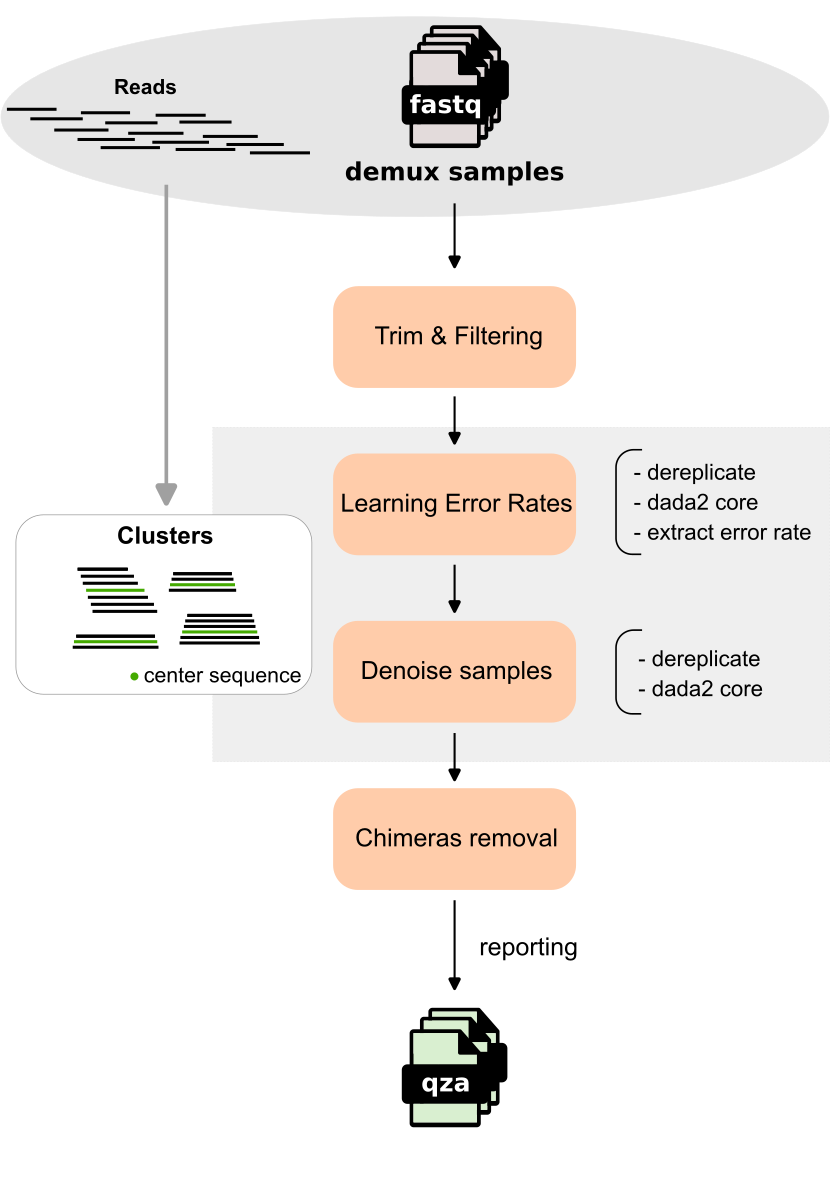
The workflow begins with demultiplexed FASTQ samples undergoing Trim & Filtering to remove low-quality data, followed by Learning Error Rates to estimate error rate between unique sequence and their partition center sequence (the most abundant in a variant group).
The process then enters the Denoise samples stage where a specialized algorithm performs constructing priors from unique sequences, and calculating -values to distinguish and remove sequencing noise from biological variants.
Once the samples are denoised, the pipeline handles Chimeras Removal to eliminate chimeras (unique sequence belongs two samples, originating from PCR) before reporting the final results as artifacts for downstream analysis.
Trim & Filtering¶
Demutiplexed files are filtered simultaneously according to following quality filtering criteria:
| Criterion | Value | Description |
|---|---|---|
truncLen | 120 | Truncates reads at 120 bases. Reads shorter than this are discarded. |
truncQ | 2 | Truncates reads at the first instance of a quality score 2. |
maxEE | 2 | Discards reads with > 2 expected errors after truncation. |
maxN | 0 | Discards sequences containing any ambiguous bases (Ns). |
minLen | 20 | Minimum read length required after trimming/truncation. |
trimLeft | 0 | No nucleotides removed from the start of the read. |
trimRight | 0 | No nucleotides removed from the end of the read. |
maxLen | Inf | No maximum length limit enforced on raw reads. |
minQ | 0 | No specific minimum quality score threshold enforced (beyond truncQ). |
rm.phix | TRUE | Discards reads matching the phiX genome. |
The values of trimLeft and truncLen are inherited from the command. Criteria descriptions adapted from dada2::fastqFilter documentation (function handles trim and filtering directly).
DADA2 core¶
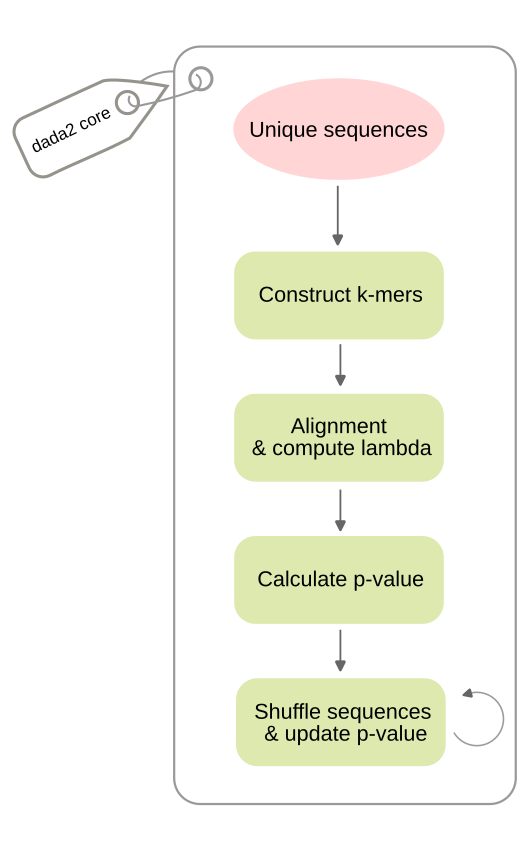
The DADA2 core algorithm (named “The divisive partitioning algorithm”) Callahan et al., 2016 begins by processing unique sequences obtained through dereplication, maintaining their respective abundance counts. To optimize the alignment step, the algorithm constructs k-mers and compute distance of pair of sequences to screen out unrelated pairs Sun et al., 2009.
During the Alignment & Compute phase, this k-mer distance acts as a pre-filter before the banded Needleman-Wunsch algorithm Gibrat, 2018 performs ends-free global pairwise alignments. From these alignments, the error rate () is calculated as the joint probability of the observed sequence originating from the partition center sequence over the total sequence length . This transition probability is determined by the specific nucleotide substitution and the associated quality score at each position .
Under the assumption that sequencing errors occur independently across reads, where any read from a true sequence can potentially be misread as sequence . The abundance -value is calculated as the probability of observing an abundance or greater, given the expected error count . This value is computed using a Poisson distribution, normalized by the probability of observing sequence at least once.
A lower -value indicates a lower probability that the observed abundance of sequence can be explained by stochastic sequencing errors originating from sequence . Therefore, a small -value provides strong evidence that sequence is a distinct biological variant.
Once the -values are computed, at the Shuffle sequences and update -value step, any sequence with a -value below the threshold () triggers the formation of a new partition with that sequence as its center.
The algorithm then enters an iterative refinement loop: -values for all unique sequences are re-calculated against the new set of centers, and sequences are reshuffled (reassigned) to the partitions for which they have the highest likelihood (most probable origin). This cycle of partitioning and reshuffling continues until the composition of the partitions remains stable and none of -value violates the threshold.
Learning Error Rates¶
The process of learning error rates begins with dereplication to collapse redundant reads into unique sequences. Next, the algorithm computes and extracts error rates by executing the DADA2 core algorithm through the “Calculate -value” step. Notably, the iterative refining loop is omitted during this phase.
Denoise samples¶
The denoising step utilizes the error rates estimated in the previous stage and performs dereplication, and the DADA2 core algorithm again. This phase focuses on detecting new center sequences (ASVs) and stabilizing partitions through the final iterative refinement loop, ensuring each sequence is assigned to its most likely biological origin.
Chimeras Removal¶
Output from denosing including center sequences (ASVs) and associated with their abundance values, from this a sequence table is compiled where columns represent unique sequences, and rows represent the samples (in our case is from 1 to 34), with the cells filled by abundance values.
When running in consensus mode, bimera identification is treated as a voting process across samples. A sequence is flagged within each individual sample where it appears; if the sequence is flagged in all samples, or in a sufficiently high fraction of them (determined by a threshold), it is definitively identified as a bimera and removed.
Summary¶
DADA2 operates on the fundamental assumption that abundance of error following Poisson distribution. A unique sequence is only promoted to a new ASV if its abundance is significantly higher than the expected error rate of its parent “center” sequence.
The DADA2 Core algorithm is applied separately among samples with two phases. Currently the algorithm run twice, maybe in future it could be restructured to perform for specific task without overlapping the each other (-value is computed uneccessarily by Learning Error Rates).
The accuracy of the Chimeras Removal step is directly enhanced by the number of samples processed. As the sample count increases, the statistical power of “vote” grows. More samples provide a higher probability of detecting the sequence which has real biological meaning.
- Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., & Holmes, S. P. (2016). DADA2: High-resolution Sample Inference from Illumina Amplicon Data. Nature Methods, 13(7), 581–583. 10.1038/nmeth.3869
- Nearing, J. T., Douglas, G. M., Comeau, A. M., & Langille, M. G. I. (2018). Denoising the Denoisers: An Independent Evaluation of Microbiome Sequence Error-Correction Approaches. PeerJ, 6, e5364. 10.7717/peerj.5364
- Sun, Y., Cai, Y., Liu, L., Yu, F., Farrell, M. L., McKendree, W., & Farmerie, W. (2009). ESPRIT: Estimating Species Richness Using Large Collections of 16S rRNA Pyrosequences. Nucleic Acids Research, 37(10), e76–e76. 10.1093/nar/gkp285
- Gibrat, J.-F. (2018). A Short Note on Dynamic Programming in a Band. BMC Bioinformatics, 19(1), 226. 10.1186/s12859-018-2228-9